




Contents
Scroll to:
https://doi.org/10.23947/2687-1653-2026-26-1-2283
EDN: IAAMVK
Scroll to:
Introduction. Modern quantum and optoelectronics, as well as nonlinear optics, place high demands on the physical and chemical properties of the materials used. This necessitates, among other things, the search for new materials that possess the properties required for a given application. At the same time, this approach can complicate the composition and crystal structure of the resulting compounds. The electronic structure of complex compounds determines their electrical, optical, magnetic, and chemical properties. These properties are unique to each compound. However, it is known that different compounds that are similar in some important parameters, for example isoelectronic ones, exhibit similarities in the structure of their electronic shells. The accumulation of such information on individual compounds and their groups necessitates generalizing the data obtained. The research objective is to consider some general characteristics of the electronic structure exhibited by groups of different compounds (chalcogenides, halides, and oxides).
Materials and Methods. The subject of study was three groups of compounds: chalcogenides Tl3TaS4, Tl3PS4, Sn2P2S6, InPS4, Cu2CdGeS4, Ag2CdSnS4, Ag2HgSnS4, halides Cs2HgX4 (X = Cl, Br, I), group APb2Br5 (A = K, Rb), and oxides La2Zr2O7, Nd2Zr2O7, Sm2Zr2O7, Eu2Zr2O7, Gd2Zr2O7. The research method involved quantum-mechanical calculations within the framework of density functional theory with various exchange-correlation potentials. Potentials were used that allowed for strong correlations between d- and f-electrons and yield a band gap value close to the experimental value.
Results. Quantum-mechanical calculations of the electronic state densities and optical characteristics of a number of chalcogenides, halides, and oxides were performed. Partial and total electron densities of states (DOS) were presented. The total density of states was compared with experimental X-ray photoelectron spectra (XPS). The validity of the calculation results was confirmed. The top of the valence band was formed by the p-states of the most electronegative elements (S, Se, Te, Br, O), whereas the bottom of the valence band was formed by the s-states of these same electronegative elements.
Discussion. Based on the calculations, general conclusions were drawn regarding the similarities in the valence band structure of the compounds considered. Using the compound Tl3TaS4 as an example, it was shown that in a solid, compared to the energies in a free atom, the binding energy of the levels for electronegative elements was significantly reduced, while for electropositive elements, it was increased. A rare-earth element (using Eu2Zr2O7 as an example) significantly altered the electron-energy structure, such that the electron states of the rare-earth element (4f-, 5p-) and the 5s-states of europium (Eu) altered the structure of the valence band of pyrochlore (Eu2Zr2O7). The calculated total and partial DOS were compared with experimental X-ray and X-ray photoelectron spectra, which confirmed the accuracy of the calculations. However, the calculated DOS curves contained numerous fine-structure elements that were obscured by instrumental distortion in the experimental curves. Thus, the calculation complemented the experiment very well, providing a more detailed picture of the electron-energy structure of the studied compounds.
Conclusion. The research objective was achieved: some general characteristics of the electronic structure exhibited by groups of different compounds (chalcogenides, halides, and oxides) were examined. The problems of identifying the states that determined the features of the electronic structure and optical characteristics of the studied groups of compounds were solved. This research can be used in the modeling of new materials with desired properties.
Lavrentyev A.A., Gabrelian B.V., Tuan V.V., Kalmykova K.F. Electronic Structure Characteristics of Complex Chalcogenides, Halides, and Oxides from Quantum-Mechanical Calculations. Advanced Engineering Research (Rostov-on-Don). 2026;26(1):2283. https://doi.org/10.23947/2687-1653-2026-26-1-2283. EDN: IAAMVK
Introduction. The tasks of semiconductor materials science include obtaining reliable information on the electron-energy structure and chemical bonding, as well as the optical characteristics of complex semiconductors, based on quantum-mechanical calculations and experimental X-ray and X-ray electron spectra. The advantage of using computational models is the ability to study hypothetical, not yet synthesized [1] compounds, as well as the elimination of the need to create suitable samples before starting the research. The objective of this work is to summarize the results of studies of various groups of compounds using reliable, widely validated computational models. Various exchange-correlation potentials were applied in the calculations, but the results closest to the experimental data were previously obtained using the modified Beke-Johnson (mBJ) potential of Tran and Blaha, with the addition of the Hubbard U correction for the strong interaction of electrons in the d- and f-shells, as well as taking into account the spin-orbital coupling (SOC), specifically for the shells of heavy elements. All references to the original articles of the authors of the approximations made in the calculations can be found in [1–4].
A comprehensive literature review of the properties and all studies of the electronic structure using both experimental and theoretical calculation methods was conducted for all the compounds studied. The review has shown the following. The Tl3TaS4 compound belongs to the materials used in devices based on the application of surface acoustic waves (SAW) [5]. It is widely used in nonlinear optics and communication devices, such as mobile telephone and television communications [6]. To meet modern technological requirements, research on improving SAW materials should be continued. Several experiments have been previously conducted to study the optical absorption edge, electronic structure, and chemical bonding features in Tl3TaS4 [7]. However, the accuracy of the obtained experimental values of the optical band gap and the absorption coefficient is significantly affected by the difficulties of observing d-orbitals over a wide energy range and a number of external factors [8]. The electronic structure and optical properties of Tl3TaS4 can be studied in detail based on ab initio calculations, which have been intensively developed and have proven their ability to reproduce reliable properties of materials [9]. The Eu2Zr2O7 compound belongs to the group of pyrochlores with the general formula A2B2O7, where A and B are metal cations, which can be trivalent (Eu) and tetravalent (Zr) [10]. Pyrochlores have significant dielectric constant. They exhibit unique magnetic [11], chemical, mechanical, and electrical properties [12]. Due to this, they can be used as ceramic coatings for thermal barriers, gas sensors, metal oxide transistors, solid electrolytes in toxic cells [13], as immobilization carriers of actinides in nuclear waste, and catalysts for oxidation reactions [14]. In order to create more efficient, reliable and functional devices based on pyrochlores [15], studies were conducted taking into account the development of new technologies in the laser engineering, optics, and materials science [15]. Calculations of the electron-energy structure of various pyrochlores were also performed within the framework of the density functional theory [16], using the exchange-correlation potential in the local density approximation (LDA) and the generalized gradient approximation (GGA). In [16], the importance of considering the Hubbard U correction when calculating the energy of d- and f-states was noted. However, it was not possible to obtain in the calculations the values of the band gap width Eg, comparable with the experimental ones (the calculated values were underestimated compared to the experimental ones).
To obtain a reliable picture of the electron energy bands in the studied pyrochlore Eu2Zr2O7, and then calculate the optical characteristics of this crystal, it was required to use other approximations to the exchange-correlation potential.
Thus, judging by the data presented in the scientific literature, numerous calculations were performed, but they required improvement of the approximations used (this is especially true for exchange-correlation potentials), which is why the Becke-Johnson potential (mBJ) was used by the authors in their early works. In addition, the above-mentioned publications also provide experimental curves of X-ray and X-ray electron spectra obtained by the authors [1] and co-authors [2–4] for comparison with the calculated curves. The objective of this work is to consider the characteristics of the electronic structure exhibited by groups of different compounds (chalcogenides, halides, and oxides).
Materials and Methods. Based on the full-potential, full-electron method of augmented plane waves (Full Potential Linearized Augmented Plane Waves, FPLAPW), implemented in the Wien2k software package [17], model quantum-mechanical calculations of the electron-energy structure of the following three groups of semiconductor compounds were previously performed:
In the unit cell of the crystal, each atom was surrounded by a muffin-tin sphere (mt-sphere), resulting in its entire volume being divided into regions occupied by mt-spheres and the remaining intersphere space. The crystal potential was then calculated in both the mt-spheres and the intersphere space. The intersphere potential was calculated by the method described in [18].
The following mt-radii were used for the atoms of the compound:



(a.u. — atomic unit of length).
The exchange-correlation potential was calculated in the GGA-PBE approximation [19] or in the mBJ approximation [20]. In the Eu2Zr2O7 compound, the rare-earth element Eu had an incomplete 4f⁷ shell. To take into account the strong Coulomb interaction of 4f electrons at one site, the Hubbard-U parameter was used, which led to the exchange-correlation potential PBE + U [21] and mBJ + U [22]. As for other compounds with a 4f shell, the EES calculation in Eu2Zr2O7 was spin-polarized.
The electronic structure of compounds with valence s-, p-, d-electrons was discussed further using the example of the Tl3TaS4 compound, crystallizing in a cubic structure with the space group I-43m, and the lattice parameter value a = 7.67 Å [1].
The crystal structure of all studied pyrochlores Ln2Zr2O7 (Ln = La, Nd, Sm, Eu, Gd), compounds with valence f-electrons, is the same and belongs to a cubic lattice with space group Fd3m. In the calculations for Eu2Zr2O7, the lattice parameter a = 10.5438 Å was used. The atomic coordinates [4] are given in Table 1.
Table 1
Coordinates of Atoms in Unit Cell of Compound Eu2Zr2O7
|
Atom |
Wyckoff symbols |
x/a |
y/a |
z/a |
|
63 Eu |
16d |
0.5 |
0.5 |
0.5 |
|
40 Zr |
16c |
0 |
0 |
0 |
|
8 O1 |
48f |
x = 0.33888 |
0.125 |
0.125 |
|
8 O2 |
8b |
0.375 |
0.375 |
0.375 |
Figure 1 shows the crystal structure and atomic environment of pyrochlore Eu2Zr2O7 [4].
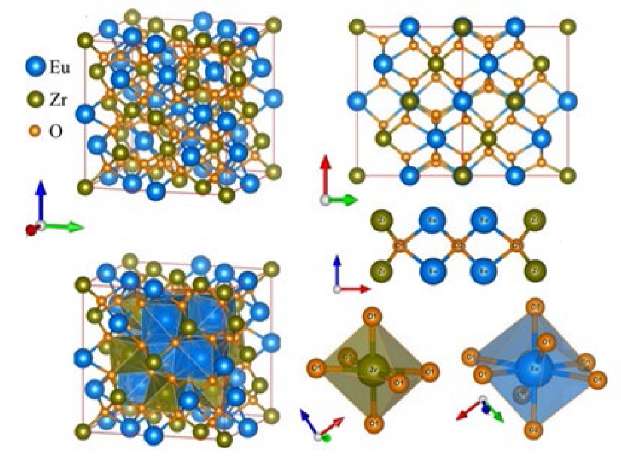
Fig. 1. Crystal structure and immediate environment of atoms in pyrochlore Eu2Zr2O7. Distances between atoms: Eu-O1 = 2.5224 Å, Eu-O2 = 2.2828 Å, Zr-O1 = 2.0858 Å
From the occupied electron states in the valence band and the free electron state in the conduction band, the combined density of states can be calculated:
 (1)
(1)
and further, using matrix elements of the transition from the valence band to the conduction band (only direct dipole transitions with Δl = 1 of the corresponding states of a single atom are considered, cross transitions, as unlikely, are ignored), the imaginary part of the permittivity tensor can be calculated [23].
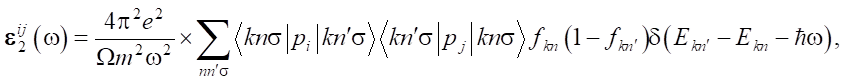 (2)
(2)
where Ekn — self-energy of the system with crystal momentum  and spin σ; m and e — electron mass and charge, respectively; V, p, |knpV⟩ and fkn — unit cell volume, momentum operator, crystal wave function, and Fermi distribution function.
and spin σ; m and e — electron mass and charge, respectively; V, p, |knpV⟩ and fkn — unit cell volume, momentum operator, crystal wave function, and Fermi distribution function.
Real part ε1(ω) of the permittivity was calculated using the Kramers-Kronig formula:
 (3)
(3)
where P — principal value of the integral.
Absorption coefficient a(ω), refractive index n(ω), extinction coefficient k(ω), optical reflection coefficient R(ω) and electron energy loss spectrum L(ω) were derived from the imaginary ε2(ω) and real ε1(ω) parts of the permittivity tensor and were calculated, respectively, from the following formulas [23].
Absorption coefficient:
 (4)
(4)
Refractive index:
 (5)
(5)
Extinction coefficient:
 (6)
(6)
Optical reflection coefficient:
 (7)
(7)
Energy loss function:
 (8)
(8)
Research Results. Electronic structure of compounds with valence s-, p-, d-states using Tl3TaS4 as an example. The results of the study on the electronic energy structure (EES) in the Tl3TaS4 compound from [1] are considered. Then, some generalizations are made on the conducted studies on the EES of the above groups of chalcogenides, halides and oxides.
Figure 2 shows a comparison of the calculated total and partial densities of electron states with the X-ray K- and L2,3 emission spectra and SK absorption spectrum.

Fig. 2. Total and partial densities of states of Tl3TaS4, calculated by mBJ+U+SOC method in comparison with experimental XPS, S Kβ1,3 and S L2,3 X-ray emission spectra and S K X-ray absorption spectrum
The value of the forbidden gap width Eg = 2.842 eV calculated in approximation mBJ+U+SOC is close to the experimental value Eg = 2.7 eV [24].
Based on the energy position of the main maxima of the X-ray emission spectra (SK- and SL2,3-bands) and the maxima of the calculated densities of electron states in the semiconductor compound Tl3TaS4 (Fig. 2), an energy diagram of the maxima of the main energy bands of this compound was constructed (Fig. 3) in comparison with the energy levels in a free atom from [25], with the latter being counted from vacuum zero. Vacuum zero is separated from the zero corresponding to the top of the valence band (Ev) in Tl3TaS4 by the work function (φ) and half the band gap (Eg). The following work function values were found for related semiconductors: for TlAsS4 and Tl3AsS3 φ = 5.5 eV, for Tl3AsS4 φ = 5.5 eV [26], which allowed us to assume value φ = 5.5 eV or Tl3TaS4 as well.
As can be seen in Figure 3, there is a symbatic decrease in the binding energy of the valence 3p and 3s levels of sulfur, the most electronegative (EN) element of the Tl3TaS4 compound (EN = 2.44). In the free atom, the binding energies of the 3p and 3s levels are – 10.36 and –20.20 eV, respectively (Table 2), while in crystal Tl3TaS4, the average values of the energies of the band maxima of these states are approximately –1.5 ÷ –2 eV for the 3p states, and –11 ÷ 12 eV for the 3s states.

Fig. 3. Energy levels in free atom [25] and maxima of experimental energy bands in Tl3TaS4 semiconductor compound relative to vacuum zero. Zero on the energy scale corresponds to the peak of valence band (Ev), EF — Fermi energy, taken at the midpoint of the band gap and at the distance of the work function from vacuum zero
In a solid, the electron density is attracted to a more electronegative atom (in this case, to sulfur S), which causes an increase in the screening of the nucleus of this atom, and therefore, as a consequence, a decrease in the binding energy of both the 3p levels and the 3s levels, compared to their values in a free atom (Table 2).
Table 2
Binding Energies of Valence and Semi-core Electrons in Free Atoms Included in Tl3TaS4 Compound (in eV) [25]
|
16 S |
3s² M1 |
3p⁴ M2 |
||
|
20.20 |
10.36 |
|||
|
73 Ta |
5d³ O4 |
5s² P1 |
||
|
8.3 |
7.9 |
|||
|
81 Tl |
5d¹⁰ O4 — O5 |
6s² P1 |
6p¹ P2 |
|
|
21 — 19 |
8 |
6.11 |
||
As stated in Blokhin's monograph [27], the effective nuclear charge is determined not only by internal electrons, but also by external electrons in relation to a given shell, that is, by all the electrons in the atom. According to these concepts, Zэфф = Z – σ1, where σ1 — total screening constant. Determining the screening constant is not the objective of this work, it is a very complex theoretical problem.
For illustration purposes, Figure 4 shows a schematic representation of the internal level shifts for positive and negative ions.

Fig. 4. Schematic representation of chemical shifts of the internal levels for positive and negative ions compared to isolated atoms: hν3 < hν1 < hν2
Indeed, for the more electropositive elements in Tl3TaS4, namely Tl and Ta, the binding energies of their valence levels increase compared to the energies of a free atom (Fig. 3 and Table 2). The screening of Tl and Tа decreases due to the electron density being drawn toward the S atom, and this causes an increase in the binding energy of the valence and semi-valence levels of the metals.
Similar conclusions about the behavior of the electronic states can be drawn for all the compounds studied (the results are published in [1–4]).
Thus, the electronic p-states of the most electronegative atoms (S, Se, Te, Br, O) in the studied chalcogenides, halides, and oxides, form the upper part of the valence band, which is associated with a significant decrease in the binding energy of these states, compared to their values in a free atom. This decrease in the binding energy of p-states can be explained by the flow of electron density to the more electronegative atom, and therefore by increased screening of these states from the nucleus. The electron s-states of the most electronegative atoms (S, Se, Te, Br, O) in chalcogenides, halides, and oxides, form the bottom of the valence band, and their binding energy also decreases compared to the energy in a free atom.
Electronic structure of compounds with valence f-states using the example of pyrochlore Eu2Zr2O7. To draw general conclusions on the electron-energy structure and optical characteristics of pyrochlores Ln2Zr2O7 (Ln = La, Nd, Sm, Eu, Gd) [3][4], the Eu2Zr2O7 compound was considered.
The valence configurations of the elements included in compound Eu2Zr2O7 are as follows:
Eu — 4f⁷5s²5p⁶6s²
Zr — 4s²4p⁶5d²5s²
O — 2s²2p⁴.
Figure 5 shows the total densities of electron states with spin-up and spin-down for two different approximations: in approximation GGA–PBE+U and GGA–PBE+U+SOC with U = 5 eV for 4f-states of Eu. The addition of spin-orbit coupling (SOC) causes a splitting of 5p⁶-states of Eu into 5p1/2 and 5p3/2-states, as well as a splitting of the 4p⁶-states Zr into 4p1/2 and 4p3/2-states.
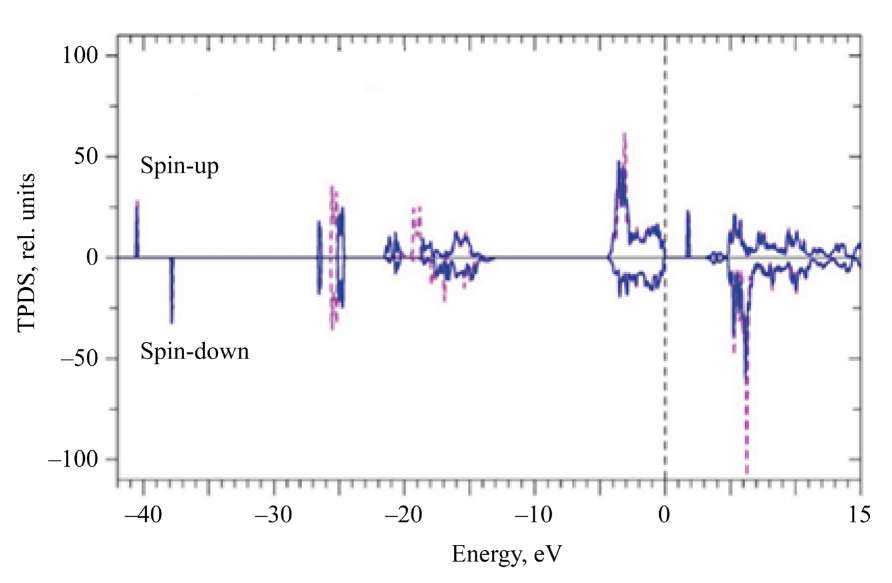
Fig. 5. Calculated total densities of electron states with spin-up and spin-down: dashed line (-----) — calculation in approximation GGA-PBE+U; solid line — calculation in approximation GGA-PBE+U+SOC; U = 5 eV for 4f- states of Eu. Zero on the energy scale corresponds to the top of the valence band Ev
The width of the forbidden band in Eu2Zr2O7, was estimated using the total densities of electron states. The data are presented in Table 3.
Table 3
Bandwidths Eg for Different Spin-up and Spin-down Calculation Approximations in Eu2Zr2O7
|
Approximation |
Eg, eV |
|
|
spin-up |
spin-down |
|
|
GGA-PBE |
0 |
0 |
|
GGA-PBE+U |
1.705 |
3.219 |
|
GGA-PBE+U+SOC |
1.667 |
|
Value of U for the 4f states of Eu is 5 eV.
The difference in the total density of electron states with spin-up and spin-down in Figure 5 can be explained using the partial densities of electron states with spin-up (Fig. 6) and spin-down (Fig. 7).

Fig. 6. Total and partial electron densities of states for spin-up in Eu2Zr2O7 calculated in approximation GGA-PBE+U+SOC. Zero energy scale corresponds to the top of EV valence band

Fig. 7. Total and partial electron densities of states for spin-down in Eu2Zr2O7 calculated in approximation GGA-PBE+U+SOC. Zero energy width corresponds to the top of valence band EV
As can be seen in Figures 6 and 7, the upper part of the valence band of Eu2Zr2O7 is formed mainly by 2p states of oxygen (region from 0 to 4.5 eV). Some admixture to the 2p states of O comes from the 4d and 5s states of Zr, as well as the 6s states of the rare-earth element Eu. The most significant admixture in the upper part of the valence band is the 4f states of Eu (the region from –2 to –4 eV). The incomplete 4f shell of Eu contains 7 electrons, whose spins align according to Hund's rule in one direction: in this calculation, this is spin-up.
The spin-orbit splitting of the 5p states of Eu can also be seen in the partial densities with spin-up and spin-down (Figs. 6 and 7). Since the 5p states of Eu are deepened relative to the 2s states of O, the interaction of these states is not as significant compared to other pyrochlores (La2Zr2O7, Nd2Zr2O7, Sm2Zr2O7).
In the present calculation, unoccupied 4f states of Eu appear in the conduction band with both spin-up (narrow peak at 2 eV) and spin-down (narrow peak in the range from 5 to 7 eV).
Another important feature, distinct from previous pyrochlores, should be noted: the peak of spin-up 4f states of Eu splits into two smaller peaks, which is due to the spin-orbit coupling of 4f⁷-electrons. These are 4f5/2- and 4f7/2-states. This is not observed in pyrochlores Nd2Zr2O7 and Sm2Zr2O7.
Despite the fact that the 4f shell is located inside the 5s, 5p, and 6s shells, i.e., it is the inner shell in the atom of a rare-earth element (Nd, Sm, Eu, Gd), the energy of the 4f states in a solid, according to the data of these calculations, falls in the upper part of the valence band, and only the 6s states are located above. This energy position of the 4f states can be explained by a significant “centrifugal” contribution to the potential energy: V(r) + l(l + 1)ℏ²/2mr², since the 4f electrons have the largest value of the orbital quantum number (l = 3).
Deeper in energy are the 2s states of O (energy range from –14.5 to –18.5 eV). Even deeper in energy are the Eu 5p states. Already in the atom, the filled 5p subshell of Eu is split due to the SOC into 5p1/2 (term О2) with an energy of 30 eV and into 5p3/2 (term О3) with an energy of 26 eV, which is reflected in Table 2. The spin-orbit splitting of the 5p states of Eu can also be seen in the partial densities with spin-up and spin-down (Figs. 6 and 7). Since the 5p states are deepened relative to the 2s states of O, the interaction of these states is no longer as significant, compared to previous pyrochlores (La2Zr2O7, Nd2Zr2O7, Sm2Zr2O7). However, in the case of Eu2Zr2O7, one can also note the admixture of the 2s states of oxygen with the 5p states of the rare-earth element Eu, which is associated with the covalency of the chemical bond Eu-O2.
The 4р⁶-states of Zr, split due to SOC, are located in the energy range of 25–27 eV. These states can be considered semi-core, not participating in the chemical bonding of Zr and O1. The semi-core 5s states of Eu are the deepest of the valence states calculated in this work. According to the Pauli exclusion principle, the two 5s electrons of Eu have different spin directions, which leads to a difference in energy between these 5s states of Eu due to the action of the magnetic field of the 4f⁷- electrons of Eu. The 5s electron of Eu with spin-up deepens to –40.5 eV, while with spin-down, it has an energy of –38 eV. The energy splitting for these states of 5s electrons is ∆E = 2.5 eV.
Calculations show that the spin-up and spin-down electron states of 5s symmetry are split in energy, which can be explained by the action of the internal magnetic field of the 4f electrons, whose spins align according to Hund's rule and act on the spin-up and spin-down 5s electrons (the Zeeman effect).
Increasing the number of 4f electrons from 4 in Nd to 6 in Sm and to 7 in Eu (Nd2Zn2O7→Sm2Zr2O7→Eu2Zr2O7) causes an increase in the magnetic field in the 4f shell and, as a consequence, an increase in the splitting of the spin-up and spin-down 5s states of the rare-earth element (Table 4).
Table 4
Magnitude of Splitting ΔE of 5s States of a Rare-Earth Element with Spin-up and Spin-down
|
Nd2Zn2O7 |
Sm2Zr2O7 |
Eu2Zr2O7 |
|
ΔE = 1.5 eV |
ΔE = 2.5 eV |
ΔE = 3.3 eV |
Figure 8 shows the total densities of electron states with spin-up and spin-down compared to the experimental X-ray photoelectron spectrum (XPS). All the fine structure features of the XPS are clearly reflected in the theoretically calculated densities of electron states. The upper part of the valence band from 0 to –5 eV is formed by the 2p states of oxygen, which is reflected in the theoretical and experimental curves.
The most intense peak А in XPS corresponds to the 4f states of Eu, since the photoionization cross section of 4f states significantly exceeds the photoionization cross section of 2p states of oxygen. At an energy of ≈–4 eV, there is an intense peak that precisely reflects the 4f states of Eu.
Gently sloping peak B in XPS corresponds mainly to the 2s states of oxygen, as well as a small fraction of the 5p3/2- states of Eu. Peak C in XPS is mainly formed by the 5p1/2- states of Eu with a small admixture of the 2s states of oxygen. Peak D in XPS corresponds to the splitting due to the SOC of the 4p states of Zr. Peak D in XPS has a clear asymmetry, which is precisely due to the spin-orbit splitting of 4р⁶- electrons into 4р1/2 and 4р3/2-states.
And finally, the wide, flat hump E on the XPS is formed by the 5s states of Eu, in which there is a splitting of the electron states with spin-up and spin-down, “smeared” on the spectrum due to hardware distortion.

Fig. 8. Total densities of electron states with spin-up and spin-down calculated in GGA-PBE+U+SOC approximation compared with experimental XPS. Zero on the energy scale corresponds to the top of the valence band Еv
Calculations of optical characteristics using Eu2Zr2O7 as an example. The output data from the EES calculations of the compound Eu2Zr2O7, namely the dispersion curves  and the DOS, were used to calculate the frequency-dependent complex dielectric function Ɛ(ω) = Ɛ1(ω) + iƐ2(ω). At the first stage of calculating the dielectric function, the imaginary part of the dielectric function tensor Ɛ2(ω) was calculated using formula (2). Figure 9 shows the imaginary part of the dielectric function Ɛ2(ω) as a function of photon energy (frequency). The main peaks and fine structure details of the curve Ɛ2(ω) are indicated: A, B, C, D, E, F, whose energies are given in Table 5.
and the DOS, were used to calculate the frequency-dependent complex dielectric function Ɛ(ω) = Ɛ1(ω) + iƐ2(ω). At the first stage of calculating the dielectric function, the imaginary part of the dielectric function tensor Ɛ2(ω) was calculated using formula (2). Figure 9 shows the imaginary part of the dielectric function Ɛ2(ω) as a function of photon energy (frequency). The main peaks and fine structure details of the curve Ɛ2(ω) are indicated: A, B, C, D, E, F, whose energies are given in Table 5.
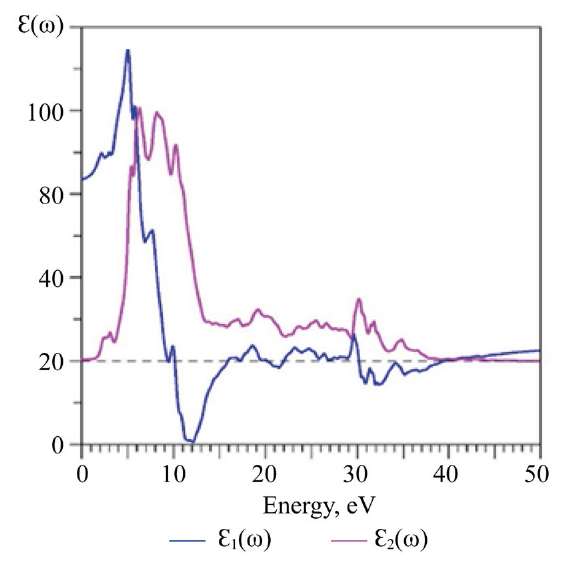
Fig. 9. Calculated imaginary (ε2) and real (ε1) parts of permittivity in Eu2Zr2O7
Together with Ɛ2(ω), Figure 9 shows a graph of the real part of the permittivity Ɛ1(ω) calculated using the Kramers–Kronig formula (3).
Table 5
Energies of Selected Peaks on Imaginary Part of Permittivity Ɛ2(ω) (Fig. 9), as Well as Reflection Coefficient R(0), Refractive Index (0) at the Beginning of Energy Count
|
A |
B |
C |
D |
E |
F |
|
5.02 eV |
8.25 |
10.16 |
19.22 |
25.62 |
30.16 |
|
Ɛ1(ω) |
8.726 |
n(0) = 2.954
R(0) = 24.423%
As in other studied pyrochlores, cross-transitions between atoms were not taken into account in the calculation of Ɛ2(ω) for Eu2Zr2O7. Thus, the key features of the fine structure of the imaginary part of the permittivity Ɛ2(ω) can be interpreted as follows:
For the Eu2Zr2O7 compound, absorption coefficient α(ω) (4), refractive index n(ω) (5), absorption coefficient k(ω) (6), optical reflection coefficient R(ω) (7) and electron energy loss spectrum L(ω) (8 were calculated, respectively, using formulas (4–8) [23]. The above optical characteristics for Eu2Zr2O7 are shown in Figures 10 and 11.

Fig. 10. Coefficients calculated in Eu2Zr2O7:
a — absorption α(ω); b — reflection R(ω)

Fig. 11. Calculated in Eu2Zr2O7:
a — refractive index n(ω) and extinction coefficient k(ω);
b — electron energy loss spectrum L(ω)
Discussion. In all groups of compounds studied in this work, the main contribution to the upper part of the valence band is given by the p-states of atoms with the highest electronegativity (chalcogens, halogens). This can be explained by a decrease in the binding energy of the p-states of chalcogens and halogens in a crystal, compared to their bonding in an isolated atom, due to the transfer of electron density from nearby metal atoms to them, and the resulting increase in the shielding of these states from the nucleus. The S-states of these atoms form the bottom of the valence band.
For compounds with atoms containing f-electrons, states of 5s-symmetry, which have different spins, are split in energy under the impact of the internal magnetic field of 4f-electrons.
The applied aspect of this research is related to the fact that the use of new materials in quantum electronics, optoelectronics, and nonlinear optics is determined by the response of the crystal under study to the action of an electron wave with a frequency lying in the optical or near and mid-IR range, that is, near the forbidden gap of the semiconductor (Eg). The crystal response can be described using the permittivity tensor (εij), which was calculated in this study for a number of complex three- and four-component chalcogenides, halides, and oxides. Of academic interest is the effect of the electron states of the rare-earth element contained in the studied pyrochlores on the electron-energy structure of these compounds.
Conclusion. The main objective of the work is achieved. The results of the study on the electron-energy structure of various groups of compounds: chalcogenides Tl3TaS4, Tl3PS4, Sn2P2S6, InPS4, Cu2CdGeS4, Ag2CdSnS4, Ag2HgSnS4, halides Cs2HgX4 (X — Cl, Br, I) and APb2Br5 (A — K, Rb), oxides La2Zr2O7 and Nd2Zr2O7 and Ln2Zr2O7 (Ln = La, Nd, Sm, Eu, Gd) are summarized. The problems of determining the effect of local partial electron states on the features of the electronic structure of the studied compounds and their optical properties are solved.
Such studies are important in the problems of modeling new materials with given properties, since they allow us to determine which factors have the main influence on the occurrence of such properties.
1. Vu Van Tuan, Lavrentyev AA, Gabrelian BV, Kalmykova KF, Sidorkin VV, Do Minh Hoat, et al. Electronic and Optical Properties of Wide Band Gap Tl3TaS4: A Promising Surface Acoustic Wave Material. Optical Materials. 2020;99:109601. https://doi.org/10.1016/j.optmat.2019.109601
2. Lavrentyev AA, Gabrelian BV, Vu Van Tuan, Khizhun OYu. Electron-Energy Structure of Complex Chalcogenides and Chalcohalogenides. Monograph. Rostov-on-Don: DSTU; 2018. 320 p. (In Russ.)
3. Lavrentyev AA, Gabrelian BV, Vu Van Tuan, Kalmykova KF. Ab initio Calculations of the Electronic-Energy Structure and Optical Properties of Lanthanum and Neodymium Pyrozirconates. Advanced Engineering Research (Rostov-on-Don). 2025;25(2):129–141. https://doi.org/10.23947/2687-1653-2025-25-2-129-141
4. Lavrentyev AA, Gabrelian BV, Vu Van Tuan, Kalmykova KF. Quantum-Mechanical Calculations of the Electronic Structure of Pyrochlores Ln2Zr2O7 (Ln = Sm, Eu, Gd) in Comparison with Experimental X-Ray Photoelectron Spectra. In: Proc. XIV International Youth Symposium “Physics of Lead-Free Piezoactive and Related Materials. Modeling of Eco-Systems”, vol II. Rostov-on-Don – Taganrog: SFU; 2025. P. 37–48.
5. Narsingh Bahadur R Singh, Ching-Hua Su, Bradley Arnold, Fow-Sen Choa, Teja Nagaradona. Effect of Impurities and Growth Parameters on the Quality of Tl3AsSe3 Optical Crystal. Optical Materials. 2016;60:81–85. https://doi.org/10.1016/j.optmat.2016.07.009
6. Bouhemadou A, Allali D, Boudiaf K, AlQarni B, Bin-Omran S, Khenata R, et al. Electronic, Optical, Elastic, Thermoelectric and Thermodynamic Properties of the Spinel Oxides ZnRh2O4 and CdRh2O4. Journal of Alloys and Compounds. 2019;774:299–314. https://doi.org/10.1016/j.jallcom.2018.09.338
7. Ewbank MD, Kowalczyk SP, Kraut EA, Harrison WA. Electronic Structure of Tl3AsSe3. Physical Review B. 1981;24(2):926–931.
8. Reshak AH. A Novel Photocatalytic Water Splitting Solar-to-Hydrogen Energy Conversion: Non-Centro-Symmetric Borate CsZn2B3O7 Photocatalyst. Journal of Alloys and Compounds. 2018;741:1258–1268. https://doi.org/10.1016/j.jallcom.2018.01.227
9. Blaha P, Schwarz K, Madsen GKH, Kvasnicka D, Luitz J, Laskowski R, et al. WIEN2k, an Augmented Plane Wave+Local Orbitals Program for Calculating Crystal Properties, rev. ed. Vienna: Vienna University of Technology; 2002. 180 p. https://pubs.aip.org/aip/jcp/article/152/7/074101/485553/WIEN2k-An-APW-lo-program-for-calculating-the
10. Chartier A, Meis C, Crocombette J, Corrales LR, Weber WJ. Atomistic Modeling of Displacement Cascades in La2Zr2O7 Pyrochlore. Physical Review B. 2003;67:174102. https://doi.org/10.1103/PhysRevB.67.174102
11. Stanek CR, Minervini L, Grimes RW. Nonstoichiometry in A2B2O7 Pyrochlores. Journal of the American Ceramic Society. 2002;85(11):2792–2798. https://doi.org/10.1111/j.1151-2916.2002.tb00530.x
12. Pirzada M, Grimes RW, Minervini L, Maguire JF, Sickafus KE. Oxygen Migration in A2B2O7 Pyrochlores. Solid State Ionics. 2001;140:201–208. https://doi.org/10.1016/S0167-2738(00)00836-5
13. Tabira Y, Withers RL, Minervini L, Grimes RW. Systematic Structural Change in Selected Rare Earth Oxide Pyrochlores as Determined by Wide-Angle CBED and a Comparison with the Results of Atomistic Computer Simulation. Journal of Solid State Chemistry. 2000;153(1):16–25. https://doi.org/10.1006/jssc.2000.8712
14. Helean KB, Ushakov SV, Brown CE, Navrotsky A, Lian J, Ewing RC, et al. Formation Enthalpies of Rare Earth Titanate Pyrochlores. Journal of Solid State Chemistry. 2004;177(6):1852–1866. https://doi.org/10.1016/j.jssc.2004.01.009
15. Chen J, Lian J, Wang LM, Ewing RC, Wang RG, Pan W. X-ray Photoelectron Spectroscopy Study of Disordering in Gd2(Ti1-xZrx)2O7 Pyrochlores. Physical Review Letters. 2002;88:105901. https://doi.org/10.1103/PhysRevLett.88.105901
16. Jing Feng, Bing Xiao, Chunlei Wan, Zheng Xiao Qu, Zheng-hong Huang, Jingchao Chen, et al. Electronic Structure, Mechanical Properties and Thermal Conductivity of Ln2Zr2O7 (Ln=La, Pr, Nd, Sm, Eu and Gd) Pyrochlore. Acta Materialia. 2011;59(4):1742–1760. https://doi.org/10.1016/j.actamat.2010.11.041
17. Blaha P, Schwarz K, Tran F, Laskowski R, Madsen GKH, Marks LD. WIEN2k: an APW+lo Program for Calculating the Properties of Solids. Journal of Chemical Physics. 2020;152(7):074101. http://users.df.uba.ar/llois/NANO/usersguide.pdf
18. Weinert M. Solution of Poisson Equation beyond Ewald-Type Method. Journal of Mathematical Physics. 1981;22(11):2433–2439. https://doi.org/10.1063/1.524800
19. Perdew JP, Burke K, Ernzerhof M. Generalized Gradient Approximation Made Simple. Physical Review Letters. 1996;77(18):3865–3869. https://doi.org/10.1103/PhysRevLett.77.3865
20. Becke AD, Johnsom ER. A Simple Effective Potential for Exchange. Journal of Chemical Physics. 2006;124(22):221101. https://doi.org/10.1063/1.2213970
21. Anisimov VI, Poteryaev AV, Korotin MA, Anokhin AO, Kotliar G. First-Principles Calculations of the Electronic Structure and Spectra of Strongly Correlated Systems: Dynamical Mean-Field Theory. Journal of Physics: Condensed Matter. 1997;9(35):7359–7367. https://doi.org/10.1088/0953-8984/9/35/010
22. Koller D, Tran F, Blaha P. Improving the Modified Becke-Johnson Exchange Potential. Physical Review B. 2012;85:155109. https://doi.org/10.1103/PhysRevB.85.155109
23. Ambrosch-Draxl C, Sofo JO. Linear Optical Properties of Solids within the Full-Potential Linearized Augmented Planewave Method. Computer Physics Communications. 2006;175(1):1–14. https://doi.org/10.1016/j.cpc.2006.03.005
24. Čermák K. Optical Absorption Edge of Tl3VS4 and Tl3TaS4. Czechoslovak Journal of Physics B. 1984;34(1):88–93. https://doi.org/10.1007/BF01590484
25. Lotz W. Electron Binding Energies in Free Atoms. Journal of the Optical Society of America. 1970:60(2):206–210. https://doi.org/10.1364/JOSA.60.000206
26. Spesivykh AA, Benz VM, Bogdanova AV. Photoemission Studies on the Energy Structure of Tl3AsS4. Russian Physics Journal. 1981;24(4):110–112. (In Russ.)
27. Blokhin MA. X-ray Physics. Moscow: Gosudarstvennoe izdatel'stvo tekhniko-teoreticheskoi literatury; 1957. 518 p. (In Russ.)
Anatoliy A. Lavrentyev, Dr.Sci. (Phys.-Math.), Professor, Head of the Electrical Engineering and Electronics Department
1, Gagarin Sq., Rostov-on-Don, 344000
Scopus Author ID: 6701574562
SPIN-code: 4121-5607
Boris V. Gabrelian, Cand.Sci. (Phys.-Math.), Associate Professor of the Computer and Automated Systems Software Department
1, Gagarin Sq., Rostov-on-Don, 344000
ResearcherID: MXM-2606-2025
Scopus Author ID: 6603244226
SPIN-code: 4411-7448
Vu Van Tuan, Cand.Sci. (Phys.-Math.), Leading Researcher at the Laboratory of Computational Physics, Institute of Computational Science and Artificial Intelligence, Van Lang University
69/68, Dang Thuy Tram, Binh Loi Trung Ward, Ho Chi Minh City
Scopus Author ID: 57214105974
Kseniya F. Kalmykova, Senior Lecturer of the Electrical Engineering and Electronics Department
1, Gagarin Sq., Rostov-on-Don, 344000
Scopus Author ID: 57211123433
SPIN-code: 6494-2333
General principles of the electronic structure of chalcogenides, halides, and oxides are studied using quantum-mechanical methods. The top of the valence band is formed by the p-states of electronegative elements. The bottom of the valence band is formed by the s-states of these same elements. Calculations have revealed fine structure details invisible in experiments. The results are applicable to modeling optoelectronic materials. The approach allows predicting the properties of new compounds of a given composition.
Lavrentyev A.A., Gabrelian B.V., Tuan V.V., Kalmykova K.F. Electronic Structure Characteristics of Complex Chalcogenides, Halides, and Oxides from Quantum-Mechanical Calculations. Advanced Engineering Research (Rostov-on-Don). 2026;26(1):2283. https://doi.org/10.23947/2687-1653-2026-26-1-2283. EDN: IAAMVK




Advanced Engineering Research (Rostov-on-Don)
ISSN 2687-1653 (Online)
Contact with: Publisher / Editorial Office of the Journal
Publisher: Don State Technical University - DSTU, Rostov-on-Don, Russia - https://donstu.ru/en/
Editor-in-Chief: Alexey N. Beskopylny, Dr.Sci. (Eng.), Professor, Vice-Rector, Don State Technical University (Rostov-on-Don, Russia)
Don State Technical University
1, Gagarin Sq., Rostov-on-Don, 344003, Russia
tel.: +7 (863) 2738-372, e-mail: vestnik@donstu.ru
16+
Processing of personal data




































